
- Anti-hestaminic & Respiratory Drugs (21)
- Anti-inflammatory Drugs (204) +-
- Baby & Mom (1362) +-
- Baby & Mom > Bath, skin & Hair > Skin Care > wibes (53)
- Beauty (3192) +-
- Beauty > Skin Care > whitening (308)
- Chemotherapy & Immune Response (891) +-
- Chemotherapy & Immune Response > ANTI-FUNGAL (11)
- Chemotherapy & Immune Response > Chemotherapeutic Agents > Hormone Antagonists >Enzyme Inhibitors (290)
- CIRCULATORY DISTURBANCE AGENTS (24)
- Diet & Fitness Products (292) +-
- DRUG AFFECTING CENTRAL NERVOUS SYSTEM (193)
- HEMATOLOGY (41)
-
Medical Supplies (533)
+-
- Chemicals & Disinfectants (19)
- Dental Supplies (38)
- Devices & Instruments (11)
- Diabetic Supplies (141)
- General Medical Supplies (21)
- I.V & Medical Solution (0)
- Intensive Care Unit & Anesthesia Supplies (0)
- KIDNEY UNIT SUPPLIES (21)
- Lab Supplies (3)
- Miscellaneous (21)
- Neonatal Unit Supplies (0)
- Operation Room Supplies (2)
- Sanitary (5)
- Sterilization Supplies (1)
- Surgical Sutures (4)
- Syringes (3)
-
Medicines & Health (2808)
+-
- Allergy & Sinus (98)
- Children's Health Care (54)
- Cough, Cold & Flu (293)
- Digestive Health & Nausea (243)
- Ear, Nose & Throat Care (189)
- Eye Care (134)
- Feminine Care (324)
- Foot Care (12)
- Orthopaedic Appliances (1)
- Pain Relief & Management (251)
- Pill Organizer (2)
- Skin Treatments (889)
- Sleep & Snoring Aids (2)
- Support & Braces (8)
- Medicines & health > Gout releif (40)
- Natural & Organic Products (82) +-
- OTC > Analgesics > Anti-inflammatory Drugs (47)
-
Personal Care (3406)
+-
- Bath & Body (277)
- Deodorant & Anti-perspirants (191)
- Ear, Nose & Throat Care (185)
- Eye Care (137)
- Feminine Care (376)
- Foot Care (21)
- Hair Care (511)
- Home Tests & Monitorings (17)
- Incontinence (7)
- Lip Care (29)
- Massage & Relaxation (17)
- Natural & Organic Personal Care (7)
- Oral Care (94)
- Pregnancy & Fertility (68)
- Shaving & Grooming (75)
- Sun Care (85)
-
Prescription Drugs (3039)
+-
- Analgesics (188)
- Cardiovascular System (391)
- Drugs Affecting Musculoskeletal System (66)
- Drugs Used In Infections (56)
- Ear & Nose Drugs (2)
- Endocrine System (209)
- Gastrointestinal Tract (255)
- Gastrointestinal Tract > Hepatology > Liver treatment (61)
- GYNECOLOGY (2)
- Miscellaneous (11)
- NEPHROLOGY > URINARY SYSTEM > RENAL DISORDERS > URINARY TRACT DISORDERS (48)
- NEUROLOGY (235)
- Nutrients & Blood Electrolytes (2)
- Respiratory System (159)
- SKIN > NAILS > HAIR > TOPICAL PREPARATIONS (126)
- Vaccines (1)
- Prescription drugs > Cardiovascular system > Anti-hypertension drugs (248)
- Sexual Wellness (317) +-
- Vitamins & Minerals Supplements (1259) +-
 solution for injection in pre-filled pen")
Availability: In Stock
Ex Tax: 2,500EGP
Example
You can return the product within 14 days of purchase.
ReturnsYou can return the product within 14 days of purchase.

Trulicity 0.75 mg solution for injection in pre-filled pen.
Trulicity 1.5 mg solution for injection in pre-filled pen.
Trulicity 0.75 mg solution for injection in pre-filled syringe.
Trulicity 1.5 mg solution for injection in pre-filled syringe.
Pre-filled pen
Trulicity 0.75 mg solution for injection
Each pre-filled pen contains 0.75 mg of dulaglutide* in 0.5 ml solution.
Trulicity 1.5 mg solution for injection
Each pre-filled pen contains 1.5 mg of dulaglutide* in 0.5 ml solution.
Pre-filled syringe
Trulicity 0.75 mg solution for injection
Each pre-filled syringe contains 0.75 mg of dulaglutide* in 0.5 ml solution.
Trulicity 1.5 mg solution for injection
Each pre-filled syringe contains 1.5 mg of dulaglutide* in 0.5 ml solution.
*Produced in CHO cells by recombinant DNA technology.
For the full list of excipients, see section 6.1.
Solution for injection.
Clear, colourless solution.
Type 2 Diabetes Mellitus
Trulicity is indicated for the treatment of adults with insufficiently controlled type 2 diabetes mellitus as an adjunct to diet and exercise
• as monotherapy when metformin is considered inappropriate due to intolerance or contraindications.
• in addition to other medicinal products for the treatment of diabetes.
For study results with respect to combinations, effects on glycaemic control and cardiovascular events, and the populations studied, see sections 4.4, 4.5 and 5.1.
Posology
Monotherapy
The recommended dose is 0.75 mg once weekly.
Add-on therapy
The recommended dose is 1.5 mg once weekly.
For potentially vulnerable populations 0.75 mg once weekly can be considered as a starting dose.
When Trulicity is added to existing metformin and/or pioglitazone therapy, the current dose of metformin and/or pioglitazone can be continued. When Trulicity is added to existing metformin and/or sodium-glucose co-transporter 2 inhibitor (SGLT2i) therapy, the current dose of metformin and/or SGLT2i can be continued. When it is added to existing therapy of a sulphonylurea or insulin, a reduction in the dose of sulphonylurea or insulin may be considered to reduce the risk of hypoglycaemia (see sections 4.4 and 4.8).
The use of Trulicity does not require blood glucose self-monitoring. Blood glucose self-monitoring is necessary to adjust the dose of sulphonylurea or insulin, particularly when Trulicity therapy is started and insulin is reduced. A stepwise approach to insulin dose reduction is recommended.
Elderly
No dose adjustment is required based on age (see section 5.2).
Renal impairment
No dosage adjustment is required in patients with mild, moderate or severe renal impairment (eGFR < 90 to ≥ 15 mL/min/1.73 m2).
There is very limited experience in patients with end stage renal disease (< 15 mL/min/1.73 m2), therefore Trulicity cannot be recommended in this population (see sections 5.1 and 5.2).
Hepatic impairment
No dosage adjustment is required in patients with hepatic impairment.
Paediatric population
The safety and efficacy of dulaglutide in children aged less than 18 years have not yet been established. No data are available.
Method of administration
Trulicity is to be injected subcutaneously in the abdomen, thigh or upper arm. It should not be administered intravenously or intramuscularly.
The dose can be administered at any time of day, with or without meals.
If a dose is missed, it should be administered as soon as possible if there are at least 3 days (72 hours) until the next scheduled dose. If less than 3 days (72 hours) remain before the next scheduled dose, the missed dose should be skipped and the next dose should be administered on the regularly scheduled day. In each case, patients can then resume their regular once weekly dosing schedule.
The day of weekly administration can be changed if necessary, as long as the last dose was administered 3 or more days (72 hours) before.
Hypersensitivity to the active substance or to any of the excipients listed in section 6.1.
Dulaglutide should not be used in patients with type 1 diabetes mellitus or for the treatment of diabetic ketoacidosis. Dulaglutide is not a substitute for insulin. Diabetic ketoacidosis has been reported in insulin-dependent patients after rapid discontinuation or dose reduction of insulin (see section 4.2).
Dehydration
Dehydration, sometimes leading to acute renal failure or worsening renal impairment, has been reported in patients treated with dulaglutide, especially at the initiation of treatment. Many of the reported adverse renal events occurred in patients who had experienced nausea, vomiting, diarrhoea, or dehydration. Patients treated with dulaglutide should be advised of the potential risk of dehydration, particularly in relation to gastrointestinal side-effects and take precautions to avoid fluid depletion.
Dulaglutide has not been studied in patients with severe gastrointestinal disease, including severe gastroparesis, and is therefore not recommended in these patients.
Acute pancreatitis
Use of GLP-1 receptor agonists has been associated with a risk of developing acute pancreatitis. In clinical trials, acute pancreatitis has been reported in association with dulaglutide (see section 4.8).
Patients should be informed of the characteristic symptoms of acute pancreatitis. If pancreatitis is suspected, dulaglutide should be discontinued. If pancreatitis is confirmed, dulaglutide should not be restarted. In the absence of other signs and symptoms of acute pancreatitis, elevations in pancreatic enzymes alone are not predictive of acute pancreatitis (see section 4.8).
Hypoglycaemia
Patients receiving dulaglutide in combination with sulphonylurea or insulin may have an increased risk of hypoglycaemia. The risk of hypoglycaemia may be lowered by a reduction in the dose of sulphonylurea or insulin (see sections 4.2 and 4.8).
Sodium content
This medicinal product contains less than 1 mmol sodium (23 mg) per 1.5 mg dose, i.e. essentially 'sodium- free'.
Dulaglutide delays gastric emptying and has the potential to impact the rate of absorption of concomitantly administered oral medicinal products. In the clinical pharmacology studies described below, dulaglutide did not affect the absorption of the orally administered medications tested to any clinically relevant degree. However, for patients receiving oral medicinal products requiring rapid gastrointestinal absorption or prolonged release formulations the potential for altered drug exposure should be considered.
Sitagliptin
Sitagliptin exposure was unaffected when coadministered with a single dose of dulaglutide. Following coadministration with 2 consecutive doses of dulaglutide, sitagliptin AUC(0-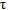 ) and Cmax decreased by approximately 7.4 % and 23.1 %, respectively. Sitagliptin tmax increased approximately 0.5 hours following coadministration with dulaglutide compared to sitagliptin alone.
) and Cmax decreased by approximately 7.4 % and 23.1 %, respectively. Sitagliptin tmax increased approximately 0.5 hours following coadministration with dulaglutide compared to sitagliptin alone.
Sitagliptin can produce up to 80 % inhibition of DPP-4 over a 24-hour period. Dulaglutide coadministration with sitagliptin increased dulaglutide exposure and Cmax by approximately 38 % and 27 %, respectively, and median tmax increased approximately 24 hours. Therefore, dulaglutide does have a high degree of protection against DPP-4 inactivation (see section 5.1, Mechanism of action). The increased exposure may enhance the effects of dulaglutide on blood glucose levels.
Paracetamol
Following a first dose of 1 and 3 mg dulaglutide, paracetamol Cmax was reduced by 36 % and 50 %, respectively, and the median tmax occurred later (3 and 4 hours, respectively). After coadministration with up to 3 mg of dulaglutide at steady state, there were no statistically significant differences on AUC(0-12), Cmax or tmax of paracetamol. No dose adjustment of paracetamol is necessary when administered with dulaglutide.
Atorvastatin
Coadministration of dulaglutide with atorvastatin decreased Cmax and AUC(0-∞) up to 70 % and 21 %, respectively, for atorvastatin and its major metabolite o-hydroxyatorvastatin. The mean t1/2 of atorvastatin and o-hydroxyatorvastatin were increased by 17 % and 41 %, respectively, following dulaglutide administration. These observations are not clinically relevant. No dose adjustment of atorvastatin is necessary when administered with dulaglutide.
Digoxin
After coadministration of steady state digoxin with 2 consecutive doses of dulaglutide, overall exposure (AUC) and tmax of digoxin were unchanged; and Cmax decreased by up to 22 %. This change is not expected to have clinical consequences. No dose adjustment is required for digoxin when administered with dulaglutide.
Anti-hypertensives
Coadministration of multiple dulaglutide doses with steady state lisinopril caused no clinically relevant changes in the AUC or Cmax of lisinopril. Statistically significant delays in lisinopril tmax of approximately 1 hour were observed on Days 3 and 24 of the study. When a single dose of dulaglutide and metoprolol were coadministered, the AUC and Cmax of metoprolol increased by19 % and 32 %, respectively. While metoprolol tmax was delayed by 1 hour, this change was not statistically significant. These changes were not clinically relevant; therefore no dose adjustment of lisinopril or metoprolol is necessary when administered with dulaglutide.
Warfarin
Following dulaglutide coadministration, S- and R-warfarin exposure and R-warfarin Cmax were unaffected, and S-warfarin Cmax decreased by 22 %. AUCINR increased by 2 %, which is unlikely to be clinically significant, and there was no effect on maximum international normalised ratio response (INRmax). The time of international normalised ratio response (tINRmax) was delayed by 6 hours, consistent with delays in tmax of approximately 4 and 6 hours for S- and R-warfarin, respectively. These changes are not clinically relevant. No dose adjustment for warfarin is necessary when given together with dulaglutide.
Oral contraceptives
Coadministration of dulaglutide with an oral contraceptive (norgestimate 0.18 mg/ethinyl estradiol 0.025 mg) did not affect the overall exposure to norelgestromin and ethinyl estradiol. Statistically significant reductions in Cmax of 26 % and 13 % and delays in tmax of 2 and 0.30 hours were observed for norelgestromin and ethinyl estradiol, respectively. These observations are not clinically relevant. No dose adjustment for oral contraceptives is required when given together with dulaglutide.
Metformin
Following coadministration of multiple dose dulaglutide with steady state metformin (immediate release formula [IR]), metformin AUC increased up to 15 % and Cmax decreased up to 12 %, respectively, with no changes in tmax. These changes are consistent with the gastric emptying delay of dulaglutide and within the pharmacokinetic variability of metformin and thus are not clinically relevant. No dose adjustment for metformin IR is recommended when given with dulaglutide.
Pregnancy
There are no or limited amount of data from the use of dulaglutide in pregnant women. Studies in animals have shown reproductive toxicity (see section 5.3). Therefore, the use of dulaglutide is not recommended during pregnancy.
Breast-feeding
It is unknown whether dulaglutide is excreted in human milk. A risk to newborns/infants cannot be excluded. Dulaglutide should not be used during breast-feeding.
Fertility
The effect of dulaglutide on fertility in humans is unknown. In the rat, there was no direct effect on mating or fertility following treatment with dulaglutide (see section 5.3).
Trulicity has no or negligible influence on the ability to drive or use machines. When it is used in combination with a sulphonylurea or insulin, patients should be advised to take precautions to avoid hypoglycaemia while driving and using machines (see section 4.4).
Summary of safety profile
In the completed phase II and phase III initial registration studies, 4,006 patients were exposed to dulaglutide alone or in combination with other glucose lowering medicinal products. The most frequently reported adverse reactions in clinical trials were gastrointestinal, including nausea, vomiting and diarrhoea. In general these reactions were mild or moderate in severity and transient in nature. Results from the long-term cardiovascular outcome study with 4949 patients randomised to dulaglutide and followed for a median of 5.4 years were consistent with these findings.
Tabulated list of adverse reactions
The following adverse reactions have been identified based on evaluation of the full duration of the phase II and phase III clinical studies, the long-term cardiovascular outcome study and post-marketing reports. The adverse reactions are listed in Table 1 as MedDRA preferred term by system organ class and in order of decreasing incidence (very common: ≥ 1/10; common: ≥ 1/100 to < 1/10; uncommon: ≥ 1/1,000 to < 1/100; rare: ≥ 1/10,000 to < 1/1,000; very rare: < 1/10,000 and not known: cannot be estimated from available data). Within each incidence grouping, adverse reactions are presented in order of decreasing frequency. Frequencies for events have been calculated based on their incidence in the phase II and phase III registration studies.
Table 1: The frequency of adverse reactions of dulaglutide
|
System Organ Class |
Very common |
Common |
Uncommon |
Rare |
Not Known |
|
Immune system disorders |
|
|
Hypersensitivity |
Anaphylactic reaction# |
|
|
Metabolism and nutrition disorders |
Hypoglycaemia* (when used in combination with insulin, glimepiride, metformin† or metformin plus glimepiride) |
Hypoglycaemia* (when used as monotherapy or in combination with metformin plus pioglitazone) |
Dehydration |
|
|
|
Gastrointestinal disorders |
Nausea, diarrhoea, vomiting†, abdominal pain† |
Decreased appetite, dyspepsia, constipation, flatulence, abdominal distention, gastroesophageal reflux disease, eructation |
|
Acute pancreatitis |
Non-mechanical intestinal obstruction |
|
Hepatobiliary disorders |
|
|
Cholelithiasis, cholecystitis |
|
|
|
Skin and subcutaneous tissue disorders |
|
|
|
Angioedema# |
|
|
General disorders and administration site conditions |
|
Fatigue |
Injection site reactions |
|
|
|
Investigations |
|
Sinus tachycardia, first degree atrioventricular block (AVB) |
|
|
|
# From post-marketing reports.
* Documented, symptomatic hypoglycaemia with blood glucose ≤ 3.9 mmol/L
† Dulaglutide 1.5 mg dose only. For dulaglutide 0.75 mg, adverse reaction met frequency for next lower incidence grouping.
Description of selected adverse reactions
Hypoglycaemia
When dulaglutide 0.75 mg and 1.5 mg were used as monotherapy or in combination with metformin alone or metformin and pioglitazone, the incidences of documented symptomatic hypoglycaemia were 5.9% to 10.9% and the rates were 0.14 to 0.62 events/patient/year, and no episodes of severe hypoglycaemia were reported.
The incidences of documented symptomatic hypoglycaemia when dulaglutide 0.75 mg and 1.5 mg, respectively, were used in combination with a sulphonylurea and metformin were 39.0% and 40.3% and the rates were 1.67 and 1.67 events/patient/year. The severe hypoglycaemia event incidences were 0% and 0.7%, and rates were 0.00 and 0.01 events/patient/year for each dose, respectively. The incidence of documented symptomatic hypoglycaemia when dulaglutide 1.5 mg was used with sulphonylurea alone was 11.3% and the rate was 0.90 events/patient/year, and there were no episodes of severe hypoglycaemia.
The incidence of documented symptomatic hypoglycaemia when dulaglutide 1.5 mg was used in combination with insulin glargine was 35.3% and the rate was 3.38 events/patient/year. The severe hypoglycaemia event incidence was 0.7% and the rate was 0.01 events/patient/year.
The incidences when dulaglutide 0.75 mg and 1.5 mg, respectively, were used in combination with prandial insulin were 85.3% and 80.0% and rates were 35.66 and 31.06 events/patient/year. The severe hypoglycaemia event incidences were 2.4% and 3.4%, and rates were 0.05 and 0.06 events/patient/year.
Gastrointestinal adverse reactions
Cumulative reporting of gastrointestinal events up to 104 weeks with dulaglutide 0.75mg and 1.5 mg, respectively, included nausea (12.9% and 21.2 %), diarrhoea (10.7% and 13.7 %) and vomiting (6.9% and 11.5 %). These were typically mild or moderate in severity and were reported to peak during the first 2 weeks of treatment and rapidly declined over the next 4 weeks, after which the rate remained relatively constant.
In clinical pharmacology studies conducted in patients with type 2 diabetes mellitus up to 6 weeks, the majority of gastrointestinal events were reported during the first 2-3 days after the initial dose and declined with subsequent doses.
Acute pancreatitis
The incidence of acute pancreatitis in phase II and III clinical studies was 0.07% for dulaglutide compared to 0.14% for placebo and 0.19% for comparators with or without additional background antidiabetic therapy.
Pancreatic enzymes
Dulaglutide is associated with mean increases from baseline in pancreatic enzymes (lipase and/or pancreatic amylase) of 11 % to 21 % (see section 4.4). In the absence of other signs and symptoms of acute pancreatitis, elevations in pancreatic enzymes alone are not predictive of acute pancreatitis.
Heart rate increase
Small mean increases in heart rate of 2 to 4 beats per minute (bpm) and a 1.3% and 1.4 % incidence of sinus tachycardia, with a concomitant increase from baseline ≥ 15 bpm, were observed with dulaglutide 0.75mg and 1.5 mg, respectively.
First degree AV block/PR interval prolongation
Small mean increases from baseline in PR interval of 2 to 3 msec and a 1.5% and 2.4 % incidence of first-degree AV block were observed with dulaglutide 0.75 mg and 1.5 mg, respectively.
Immunogenicity
In clinical studies, treatment with dulaglutide was associated with a 1.6 % incidence of treatment emergent dulaglutide anti-drug antibodies, indicating that the structural modifications in the GLP-1 and modified IgG4 parts of the dulaglutide molecule, together with high homology with native GLP-1 and native IgG4, minimise the risk of immune response against dulaglutide. Patients with dulaglutide anti-drug antibodies generally had low titres, and although the number of patients developing dulaglutide anti-drug antibodies was low, examination of the phase III data revealed no clear impact of dulaglutide anti-drug antibodies on changes in HbA1c. None of the patients with systemic hypersensitivity developed dulaglutide anti-drug antibodies.
Hypersensitivity
In the phase II and phase III clinical studies, systemic hypersensitivity events (e.g., urticaria, oedema) were reported in 0.5 % of patients receiving dulaglutide. Cases of anaphylactic reaction have been rarely reported with marketed use of dulaglutide.
Injection site reactions
Injection site adverse events were reported in 1.9 % of patients receiving dulaglutide. Potentially immune-mediated injection site adverse events (e.g., rash, erythema) were reported in 0.7 % of patients and were usually mild.
Discontinuation due to an adverse event
In studies of 26 weeks duration, the incidence of discontinuation due to adverse events was 2.6% (0.75 mg) and 6.1% (1.5 mg) for dulaglutide versus 3.7 % for placebo. Through the full study duration (up to 104 weeks), the incidence of discontinuation due to adverse events was 5.1% (0.75 mg) and 8.4 % (1.5 mg) for dulaglutide. The most frequent adverse reactions leading to discontinuation for 0.75 mg and 1.5 mg dulaglutide, respectively, were nausea (1.0%, 1.9 %), diarrhoea (0.5%, 0.6 %), and vomiting (0.4%, 0.6 %), and were generally reported within the first 4-6 weeks.
Reporting of suspected adverse reactions
Reporting suspected adverse reactions after authorisation of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via Ireland: HPRA Pharmacovigilance; website: www.hpra.ie, or United Kingdom: Yellow Card Scheme; website: www.mhra.gov.uk/yellowcard or search for MHRA Yellow Card in the Google Play or Apple App Store.
Effects of overdose with dulaglutide in clinical studies have included gastrointestinal disorders and hypoglycaemia. In the event of overdose, appropriate supportive treatment should be initiated according to the patient's clinical signs and symptoms.
Write a review
Your Name:Your Review: Note: HTML is not translated!
Rating: Bad Good
Enter the code in the box below:



