- Anti-hestaminic & Respiratory Drugs (21)
- Anti-inflammatory Drugs (181) +-
- Baby & Mom (1314) +-
- Baby & Mom > Bath, skin & Hair > Skin Care > wibes (53)
- Beauty (2723) +-
- Beauty > Skin Care > whitening (273)
- Chemotherapy & Immune Response (659) +-
- Chemotherapy & Immune Response > ANTI-FUNGAL (6)
- Chemotherapy & Immune Response > Chemotherapeutic Agents > Hormone Antagonists >Enzyme Inhibitors (235)
- CIRCULATORY DISTURBANCE AGENTS (23)
- Diet & Fitness Products (248) +-
- DRUG AFFECTING CENTRAL NERVOUS SYSTEM (183)
- Drugs affecting CNS >Anti- epileptic (78)
- HEMATOLOGY (12)
-
Medical Supplies (468)
+-
- Chemicals & Disinfectants (19)
- Dental Supplies (27)
- Devices & Instruments (8)
- Diabetic Supplies (107)
- General Medical Supplies (21)
- I.V & Medical Solution (0)
- Intensive Care Unit & Anesthesia Supplies (0)
- Kindney Unit Supplies (12)
- Lab Supplies (1)
- Miscellaneous (21)
- Neonatal Unit Supplies (0)
- Operation Room Supplies (2)
- Sanitary (5)
- Sterilization Supplies (0)
- Surgical Sutures (4)
- Syringes (3)
-
Medicines & Health (2535)
+-
- Allergy & Sinus (93)
- Children's Health Care (52)
- Cough, Cold & Flu (297)
- Digestive Health & Nausea (218)
- Ear, Nose & Throat Care (174)
- Eye Care (117)
- Feminine Care (315)
- Foot Care (4)
- Orthopaedic Appliances (0)
- Pain Relief & Management (227)
- Pill Organizer (2)
- Skin Treatments (734)
- Sleep & Snoring Aids (0)
- Support & Braces (6)
- Medicines & health > Gout releif (42)
- Natural & Organic Products (82) +-
- OTC > Analgesics > Anti-inflammatory Drugs (43)
-
Personal Care (3044)
+-
- Bath & Body (256)
- Deodorant & Anti-perspirants (179)
- Ear, Nose & Throat Care (169)
- Eye Care (123)
- Feminine Care (362)
- Foot Care (12)
- Hair Care (388)
- Home Tests & Monitorings (14)
- Incontinence (7)
- Lip Care (20)
- Massage & Relaxation (18)
- Natural & Organic Personal Care (7)
- Oral Care (81)
- Pregnancy & Fertility (60)
- Shaving & Grooming (67)
- Sun Care (67)
- Prescribtion drugs > cardiovascular system > Hypertention drugs (334) +-
-
Prescription Drugs (2988)
+-
- Analgesics (180)
- Cardiovascular System (347)
- Drugs Affecting Musculoskeletal System (62)
- Drugs Used In Infections (53)
- Ear & Nose Drugs (2)
- Endocrine System (157)
- Gastrointestinal Tract (232)
- Gastrointestinal Tract (214)
- GYNECOLOGY (2)
- Miscellaneous (6)
- NEPHROLOGY > URINARY SYSTEM > RENAL DISORDERS > URINARY TRACT DISORDERS (24)
- NEUROLOGY (210)
- Nutrients & Blood Electrolytes (2)
- prescription drugs > cardiovascular system >Anti-hypertensive drugs (78)
- Prescription Drugs > Gastrointestinal Tract > Hepatology > Liver treatment (57)
- Respiratory System (137)
- SKIN > NAILS > HAIR > TOPICAL PREPARATIONS (42)
- Vaccines (1)
- Sexual Wellness (258) +-
- strong anti-emetic & adjuvent used with anti-neoplastic (0)
- Vitamins & Minerals Supplements (1134) +-
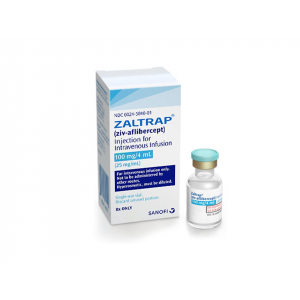
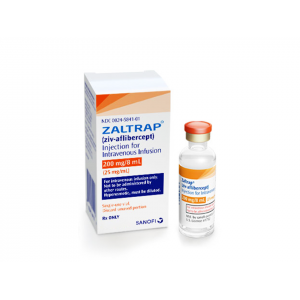
 INJECTION FOR INTRAVENOUS INFUSION VIAL 8 ML")
Ex Tax: 9,680EGP
Example
You can return the product within 14 days of purchase.
ReturnsYou can return the product within 14 days of purchase.

ZALTRAP (ZIV-AFLIBERCEPT)
INDICATION
ZALTRAP®, in combination with fluorouracil, leucovorin, irinotecan-(FOLFIRI), is indicated for the treatment of patients with metastatic colorectal cancer (mCRC) that is resistant to or has progressed following an oxaliplatin-containing regimen.
IMPORTANT SAFETY INFORMATION
WARNINGS AND PRECAUTIONS
Hemorrhage: Patients treated with ZALTRAP have an increased risk of hemorrhage, including severe and sometimes fatal hemorrhagic events.
Bleeding/hemorrhage (all grades) was reported in 38% of ZALTRAP/FOLFIRI patients vs. 19% of placebo/FOLFIRI patients. Grade 3-4 hemorrhagic events, including GI hemorrhage, hematuria, and post-procedural hemorrhage, occurred in 3% of ZALTRAP/FOLFIRI patients vs. 1% of placebo/FOLFIRI patients. Severe intracranial hemorrhage and pulmonary hemorrhage/hemoptysis including fatal events have occurred in patients receiving ZALTRAP.
Monitor patients for signs and symptoms of bleeding. Do not initiate ZALTRAP in patients with severe hemorrhage. Discontinue ZALTRAP in patients who develop severe hemorrhage.
Gastrointestinal Perforation: GI perforation including fatal GI perforation can occur in patients receiving ZALTRAP.
Across three clinical trials (colorectal, pancreatic, and lung cancer), GI perforation (all grades/Grade 3-4) occurred in 0.8% /0.8% of Zaltrap patients and 0.3% /0.2% for placebo patients.
Monitor patients for signs and symptoms of GI perforation. Discontinue ZALTRAP in patients who experience GI perforation.
Impaired Wound Healing: Grade 3 impaired wound healing was reported in 2 patients (0.3%) treated with ZALTRAP/FOLFIRI.
Discontinue ZALTRAP in patients with impaired wound healing. The safety of resumption of ZALTRAP after resolution of wound healing complications has not been established.
Withhold ZALTRAP for at least 4 weeks prior to elective surgery and do not administer ZALTRAP for at least 4 weeks after major surgery and until wounds have adequately healed.
For minor surgery such as central venous access port placement, biopsy, and tooth extraction, ZALTRAP may be initiated/resumed once the surgical wound is fully healed.
Fistula Formation: Fistula formation involving GI and non-GI sites occurs at a higher incidence in patients treated with ZALTRAP. Fistulas (anal, enterovesical, enterocutaneous, colovaginal, intestinal sites) were reported in 1.5% (9/611) of ZALTRAP/FOLFIRI treated patients and 0.5% (3/605) of placebo/FOLFIRI patients. Grade 3 GI fistula formation occurred in 2 patients treated with Zaltrap (0.3%) and 1 patient treated with placebo (0.2%). Discontinue ZALTRAP therapy in patients who develop fistula.
Hypertension: An increased risk of Grade 3-4 hypertension has been observed in patients receiving ZALTRAP.
There is no clinical trial experience administering ZALTRAP to patients with NYHA class III or IV heart failure. In patients with mCRC, Grade 3 hypertension (defined as requiring adjustment in existing antihypertensive therapy or treatment with more than one drug) was reported in 1.5% of patients treated with placebo/FOLFIRI and 19% treated with ZALTRAP/FOLFIRI. Grade 4 hypertension (hypertensive crisis) was reported in 1 patient (0.2%) treated with ZALTRAP/FOLFIRI. Of patients treated with ZALTRAP/FOLFIRI who developed Grade 3-4 hypertension, 54% had onset during the first two cycles of treatment.
Monitor blood pressure at least every two weeks, treat with appropriate antihypertensive therapy, and continue monitoring blood pressure regularly during ZALTRAP treatment. Temporarily suspend ZALTRAP until hypertension is controlled, and permanently reduce the ZALTRAP dose to 2 mg/kg for subsequent cycles. Discontinue ZALTRAP in patients with hypertensive crisis or hypertensive encephalopathy.
Arterial Thromboembolic Events: Arterial thromboembolic events (ATE), including transient ischemic attack, cerebrovascular accident, and angina pectoris, occurred more frequently in patients who have received ZALTRAP. ATE occurred in 2.6% of ZALTRAP/FOLFIRI patients and 1.7% of placebo/FOLFIRI patients. Grade 3-4 events occurred in 11 patients (1.8%) treated with ZALTRAP/FOLFIRI and 4 patients (0.7%) treated with placebo/FOLFIRI. Discontinue ZALTRAP in patients who experience an ATE.
Proteinuria: Severe proteinuria, nephrotic syndrome, and thrombotic microangiopathy (TMA) occurred more frequently in patients treated with ZALTRAP.
Proteinuria was reported in 62% of ZALTRAP/FOLFIRI patients compared to 41% of placebo/FOLFIRI patients. Grade 3-4 proteinuria occurred in 8% of ZALTRAP/FOLFIRI patients compared to 1% of placebo/FOLFIRI patients. Nephrotic syndrome occurred in 2 patients (0.5%) treated with ZALTRAP/FOLFIRI compared to none of the patients treated with placebo/FOLFIRI. TMA was reported in 3 of 2,258 patients with cancer enrolled across completed studies.
Monitor proteinuria by urine dipstick analysis and/or urinary protein creatinine ratio (UPCR) for the development or worsening of proteinuria. Obtain a 24-hour urine collection in patients with a dipstick of ≥2+ for protein or UPCR >1
Suspend ZALTRAP when proteinuria ≥2 grams/24 hours and resume ZALTRAP when proteinuria <2 grams/24 hours.
If recurrent, suspend until proteinuria <2 grams/24 hours and then reduce ZALTRAP dose to 2 mg/kg.
Discontinue ZALTRAP if nephrotic syndrome or TMA develops.
Neutropenia and Neutropenic Complications: A higher incidence of neutropenic complications (febrile neutropenia and neutropenic infection) occurred in patients receiving ZALTRAP.
Grade 3-4 neutropenia occurred in 37% of ZALTRAP/FOLFIRI patients compared to 30% of placebo/FOLFIRI patients. Grade 3-4 febrile neutropenia occurred in 4% of ZALTRAP/FOLFIRI patients compared to 2% of placebo/FOLFIRI patients. Grade 3-4 neutropenic infection/sepsis occurred in 1.5% of ZALTRAP/FOLFIRI patients compared to 1.2% of placebo/FOLFIRI patients.
Monitor CBC with differential count at baseline and prior to initiation of each cycle of ZALTRAP. Delay administration of ZALTRAP/FOLFIRI until neutrophil count is ≥ 1.5 x 109/L.
Diarrhea and Dehydration: Incidence of severe diarrhea and dehydration is increased in patients treated with ZALTRAP/FOLFIRI.
Grade 3-4 diarrhea was reported in 19% of ZALTRAP/FOLFIRI patients compared to 8% of placebo/FOLFIRI patients. Grade 3-4 dehydration was reported in 4% of ZALTRAP/FOLFIRI patients compared to 1% of placebo/FOLFIRI patients.
The incidence of diarrhea is increased in patients ≥65 years of age compared to patients <65 years of age. Monitor closely.
Reversible Posterior Leukoencephalopathy Syndrome: RPLS (also known as posterior reversible encephalopathy syndrome) was reported in 0.5% of 3795 patients treated with ZALTRAP monotherapy or in combination with chemotherapy. Confirm diagnosis of RPLS with magnetic resonance imaging (MRI) and discontinue ZALTRAP in patients who develop RPLS. Symptoms usually resolve or improve within days, although some patients have experienced ongoing neurologic sequelae or death.
Embryo-Fetal Toxicity: Based on findings from animal studies and its mechanism of action, ZALTRAP can cause fetal harm when administered to pregnant women. Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with ZALTRAP and for 3 months following the last dose.
ADVERSE REACTIONS
The most common adverse reactions (≥20%) in the ZALTRAP/FOLFIRI arm were leukopenia, diarrhea, neutropenia, proteinuria, AST increased, stomatitis, fatigue, thrombocytopenia, ALT increased, hypertension, weight decreased, decreased appetite, epistaxis, abdominal pain, dysphonia, serum creatinine increased, and headache.
The most common Grade 3-4 adverse reactions (≥5%) in the ZALTRAP/FOLFIRI arm were neutropenia, diarrhea, hypertension, leukopenia, stomatitis, fatigue, proteinuria, and asthenia.
Infections occurred at a higher frequency in patients receiving ZALTRAP/FOLFIRI (46%, all grades; 12%, Grade 3-4) than in patients receiving placebo/FOLFIRI (33%, all grades; 7%, Grade 3-4), including urinary tract infection, nasopharyngitis, upper respiratory tract infection, pneumonia, catheter site infection, and tooth infection.
In patients with mCRC, venous thromboembolic events (VTE), consisting primarily of deep venous thrombosis and pulmonary embolism, occurred in 9% of patients treated with ZALTRAP/FOLFIRI and 7% of patients treated with placebo/FOLFIRI.
USE IN SPECIFIC POPULATIONS
Pregnancy: Based on findings from animal reproduction studies and its mechanism of action, ZALTRAP can cause fetal harm when administered to pregnant women. There is insufficient data in pregnant women exposed to ZALTRAP to assess the risks. Advise pregnant women of the potential risk to a fetus.
Lactation: There are no data on the presence of ZALTRAP in human milk, or the effects of ZALTRAP on the breastfed infant or on milk production. Because of the potential for serious adverse reactions in breastfed infants, advise women not to breastfeed during treatment with ZALTRAP and for 1 month following the last dose.
Females and Males of Reproductive Potential: Verify the pregnancy status in females of reproductive potential prior to initiating ZALTRAP. Advise female patients of reproductive potential to use effective contraception during treatment with ZALTRAP and for 3 months following the last dose. Advise female and male patients of reproductive potential that ZALTRAP may impair reproductive function and fertility.
Write a review
Your Name:Your Review: Note: HTML is not translated!
Rating: Bad Good
Enter the code in the box below: